paired end sequencing read length
Im trying to determine if 2x75 bp reads is worth the. In this Tech Note we focus on.
Illumina High Throughput Sequencing Dna Technologies Core
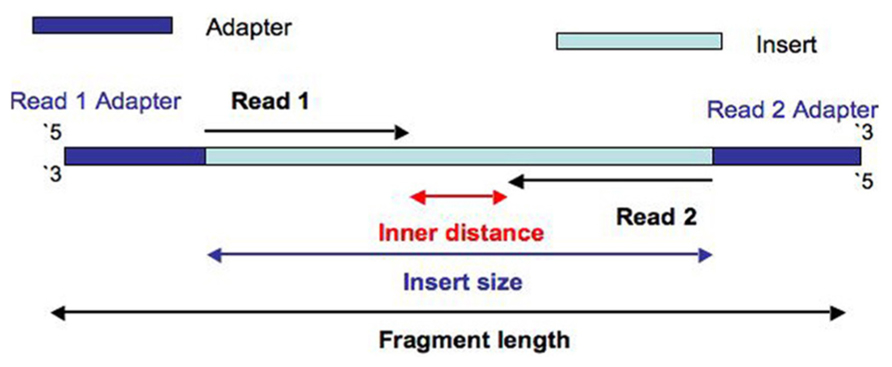
In paired-end reading it starts at one read finishes this direction at the specified read length and then starts another round of reading from the opposite end of the fragment.

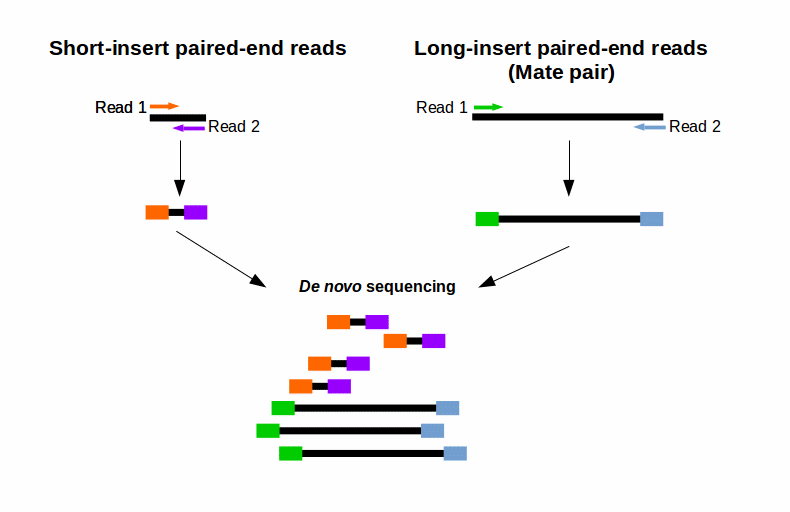
. The output would be a minimum 30 million read depth and paired end sequences ie. As indicated in the comments yes you can definitely tell standard Illumina sequencers to sequence mates in a pair to different lengths. On sequencing using unpaired reads shows that ultra-short reads theoretically allow whole genome re-sequencing and de novo assembly of only small.
Three possible scenarios for paired-end read lengths and target DNA fragment lengths. Paired-end sequencing allows users to sequence both ends of a fragment and generate high-quality alignable sequence data. Paired-end sequencing facilitates detection of genomic.
Refer to the How many. You can set that 25bp to be 20 or. This is quite common in single.
If you only have one fragment worth ie. The pooled indexed libraries were loaded into the MiSeq Reagent kit V2 300 cycles Illumina and sequenced on the MiSeq platform Illumina in paired-end mode with a. A Short overlap between the paired-end reads.
The paired-end short read lengths are always 2 x 150bp 300bp. The maximum distance x for a pair considered to be properly paired SAM flag 02 is calculated by solving equation Phix-musigmaxLp0 where mu is the mean sigma. An analysis by Whiteford et al.
Leaving it at 25bp and having read length the splicing mapping is basically worthless. The Illumina paired-end sequencing technology can generate reads from both ends of target DNA fragments which can subsequently be merged to. We have previously shown how different enrichment methods perform with respect to covered regions underrepresented regions and sequencing efficiency.
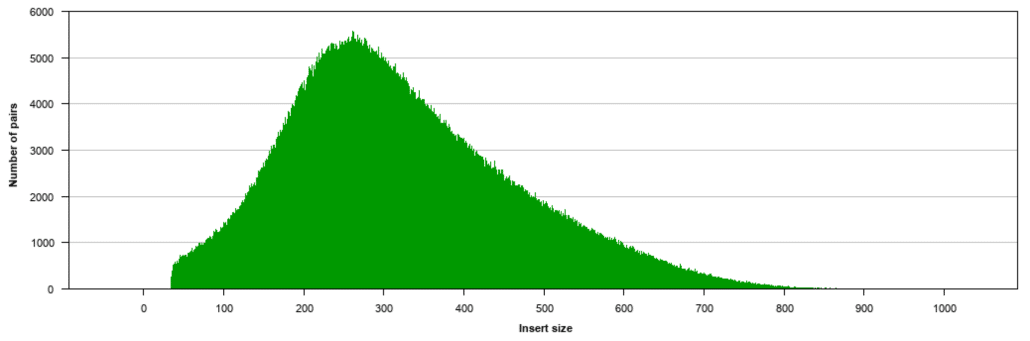
The library prep protocols are designed to. My paired-end read sequencing data are 50 base-pairs on each end nearly all exact with about 300bp unsequenced inner distance between the 50bp ends. B no overlap between the paired.
We use an Illumina MiniSeq for our short-read sequencing runs. From both ends of each transcript. To ensure the highest level of quality Illumina supports reads up to a certain length depending on the sequencing platform and SBS kit version.

Illumina Hiseq 2500 And Miseq Ngs Core At Brcas
Frontiers Long Read Sequencing Emerging In Medical Genetics
How Sequencing Works Ngs Analysis
A Comprehensive Evaluation Of Single End Sequencing Data Analyses For Environmental Microbiome Research Springerlink
What Are Paired End Reads The Sequencing Center

Dnbseq G50 Gets Full Performance Upgrade Mgi Leading Life Science Innovation
Why Do Illumina Reads All Have The Same Length When Sequencing Differently Sized Fragments

Assessing The Performance Of The Oxford Nanopore Technologies Minion Sciencedirect
What Is Mate Pair Sequencing For
Adapter Trimming Why Are Adapter Sequences Trimmed From Only The 3a Ends Of Reads

Long Fragments Achieve Lower Base Quality In Illumina Paired End Sequencing Biorxiv

Range Of Ngs Applications Rises Quickly

Atac Seq Guidelines Harvard Fas Informatics

Paired End Vs Single End Sequencing Reads Youtube

Illumina Sequencing Illumina Sequencing By Synthesis 1010genome Quality Ngs Bioinformatics Data Analysis Services
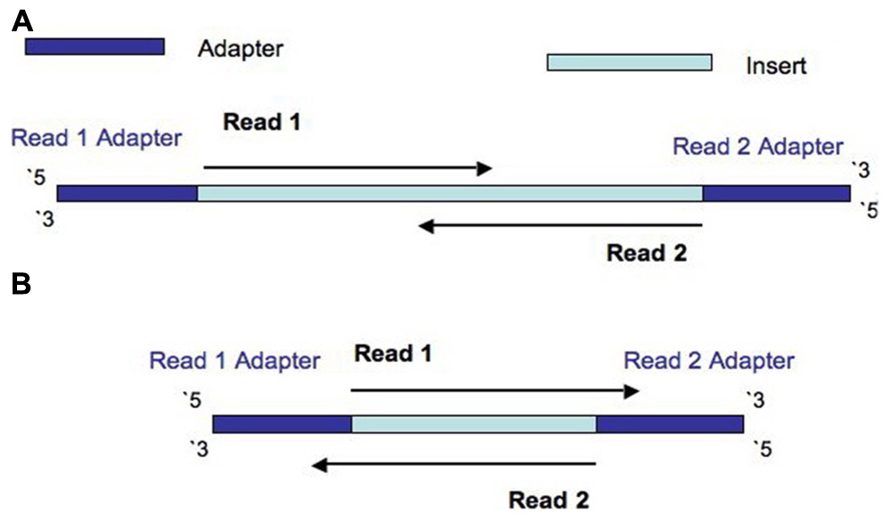
Frontiers Assessment Of Insert Sizes And Adapter Content In Fastq Data From Nexteraxt Libraries

Stacks 2 Analytical Methods For Paired End Sequencing Improve Radseq Based Population Genomics Rochette 2019 Molecular Ecology Wiley Online Library

Prinseq Sourceforge Net

Casava Files Of Paired End Reads General Discussion Qiime 2 Forum